


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Doravirine: Doravirine is a pyridinone non-nucleoside reverse transcriptase inhibitor of HIV-1 and inhibits HIV-1 replication by non-competitive inhibition of HIV-1 reverse transcriptase (RT). Doravirine does not inhibit the human cellular DNA polymerases α, β, and mitochondrial DNA polymerase γ.
Lamivudine: Lamivudine is a nucleoside analogue. Intracellularly, lamivudine is phosphorylated to its active 5'-triphosphate metabolite, lamivudine triphosphate (3TC-TP). The principal mode of action of 3TC-TP is inhibition of RT via DNA chain termination after incorporation of the nucleotide analogue.
Tenofovir disoproxil: Tenofovir disoproxil is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. Tenofovir disoproxil requires initial diester hydrolysis for conversion to tenofovir and subsequent phosphorylations by cellular enzymes to form tenofovir diphosphate. Tenofovir diphosphate inhibits the activity of HIV-1 RT by competing with the natural substrate deoxyadenosine 5'-triphosphate and, after incorporation into DNA, by DNA chain termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.
Antiviral activity in cell culture: Doravirine: Doravirine exhibited an EC50 value of 12.0±4.4 nM against wild-type laboratory strains of HIV-1 when tested in the presence of 100 % normal human serum using MT4-GFP reporter cells. Doravirine demonstrated antiviral activity against a broad panel of primary HIV-1 isolates (A, A1, AE, AG, B, BF, C, D, G, H) with EC50 values ranging from 1.2 nM to 10.0 nM. The antiviral activity of doravirine was not antagonistic when combined with lamivudine and tenofovir disoproxil.
Lamivudine: The antiviral activity of lamivudine against HIV-1 was assessed in a number of cell lines including monocytes and peripheral blood mononuclear cells (PBMCs) using standard susceptibility assays. EC50 values were in the range of 0.003 to 15 microM (1 microM = 0.23 micrograms per mL). The median EC50 values of lamivudine were 60 nM (range: 20 to 70 nM), 35 nM (range: 30 to 40 nM), 30 nM (range: 20 to 90 nM), 20 nM (range: 3 to 40 nM), 30 nM (range: 1 to 60 nM), 30 nM (range: 20 to 70 nM), 30 nM (range: 3 to 70 nM), and 30 nM (range: 20 to 90 nM) against HIV-1 clades A-G and group O viruses (n = 3 except n = 2 for clade B) respectively. Ribavirin (50 microM) used in the treatment of chronic HCV infection decreased the anti-HIV-1 activity of lamivudine by 3.5-fold in MT-4 cells.
Tenofovir disoproxil: The antiviral activity of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in T lymphoblastoid cell lines, primary monocyte/macrophage cells and peripheral blood lymphocytes. The EC50 values for tenofovir were in the range of 0.04-8.5 microM. Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC50 values ranged from 0.5-2.2 microM).
Resistance: In cell culture: Doravirine: Doravirine-resistant strains were selected in cell culture starting from wild-type HIV-1 of different origins and subtypes, as well as NNRTI-resistant HIV-1. Observed emergent amino acid substitutions in RT included: V106A, V106M, V106I, V108I, F227L, F227C, F227I, F227V, H221Y, M230I, L234I, P236L, and Y318F. The V106A, V106M, V108I, H221Y, F227C, M230I, P236L, and Y318F substitutions conferred 3.4-fold to 70-fold reductions in susceptibility to doravirine. Y318F in combination with V106A, V106M, V108I, and F227C conferred greater decreases in susceptibility to doravirine than Y318F alone, which conferred a 10-fold reduction in susceptibility to doravirine. Common NNRTI-resistant mutations (K103N, Y181C) were not selected in the in vitro study. V106A (yielding a fold change of around 19) appeared as an initial substitution in subtype B virus, and V106A or M in subtype A and C virus. Subsequently F227(L/C/V) or L234I emerged in addition to V106 substitutions (double mutants yielding a fold change of > 100).
Lamivudine: Lamivudine-resistant variants of HIV-1 have been selected in cell culture and in subjects treated with lamivudine. Genotypic analysis showed that the resistance was due to a specific amino acid substitution in the HIV-1 RT at codon 184 changing the methionine to either isoleucine or valine (M184V/I).
Tenofovir disoproxil: HIV-1 isolates selected by tenofovir expressed a K65R substitution in HIV-1 RT and showed a 2-4 fold reduction in susceptibility to tenofovir. In addition, a K70E substitution in HIV-1 RT has been selected by tenofovir and results in low-level reduced susceptibility to abacavir, emtricitabine, lamivudine, and tenofovir.
In clinical trials: Treatment-naïve adult subjects: Doravirine: The Phase 3 studies, DRIVE-FORWARD and DRIVE-AHEAD, included previously untreated patients (n = 747) where the following NNRTI substitutions were part of exclusion criteria: L100I, K101E, K101P, K103N, K103S, V106A, V106I, V106M, V108I, E138A, E138G, E138K, E138Q, E138R, V179L, Y181C, Y181I, Y181V, Y188C, Y188H, Y188L, G190A, G190S, H221Y, L234I, M230I, M230L, P225H, F227C, F227L, F227V.
The following de novo resistance was seen in the resistance analysis subset (subjects with HIV-1 RNA greater than 400 copies per mL at virologic failure or at early study discontinuation and having resistance data). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEmergent doravirine associated resistance substitutions in RT included one or more of the following: A98G, V106I, V106A, V106M/T, Y188L, H221Y, P225H, F227C, F227C/R, and Y318Y/F.
Virologically suppressed adult subjects: The DRIVE-SHIFT study included virologically suppressed patients (N = 670) with no history of treatment failure (see Clinical experience as follows). A documented absence of genotypic resistance (prior to starting first therapy) to doravirine, lamivudine, and tenofovir was part of the inclusion criteria for patients who switched from a PI- or INI-based regimen. Exclusionary NNRTI substitutions were those listed previously (DRIVE-FORWARD and DRIVE-AHEAD), with the exception of substitutions RT K103N, G190A and Y181C (accepted in DRIVE-SHIFT). Documentation of pre-treatment resistance genotyping was not required for patients who switched from a NNRTI-based regimen.
In the DRIVE-SHIFT clinical trial, no subjects developed genotypic or phenotypic resistance to DOR, 3TC, or TDF during the initial 48 weeks (immediate switch, N = 447) or 24 weeks (delayed switch, N = 209) of treatment with Delstrigo. One subject developed RT M184M/I mutation and phenotypic resistance to 3TC and FTC during treatment with their baseline regimen. None of the 24 subjects (11 in the immediate switch group, 13 in the delayed switch group) with baseline NNRTI mutations (RT K103N, G190A, or Y181C) experienced virologic failure through Week 48, or at time of discontinuation.
Paediatric subjects: In the IMPAACT 2014 (Protocol 027) clinical trial, no subject who was virologically suppressed at baseline met the criteria for resistance analysis. One treatment-naïve subject who met the protocol-defined virologic failure criteria (defined as 2 consecutive plasma HIV-1 RNA test results ≥ 200 copies/mL) at or after Week 24 was evaluated for the development of resistance; no emergence of genotypic or phenotypic resistance to doravirine, lamivudine or tenofovir was detected.
Cross-resistance: No significant cross-resistance has been demonstrated between doravirine-resistant HIV-1 variants and lamivudine/emtricitabine or tenofovir or between lamivudine- or tenofovir-resistant variants and doravirine.
Doravirine: Doravirine has been evaluated in a limited number of patients with NNRTI resistance (K103N n = 7, G190A n = 1); all patients were suppressed to < 40 copies/mL at Week 48. A breakpoint for a reduction in susceptibility, yielded by various NNRTI substitutions, that is associated with a reduction in clinical efficacy has not been established.
Laboratory strains of HIV-1 harbouring the common NNRTI-associated mutations K103N, Y181C, or K103N/Y181C substitutions in RT exhibit less than a 3-fold decrease in susceptibility to doravirine compared to wild-type virus when evaluated in the presence of 100 % normal human serum. In in vitro studies, doravirine was able to suppress the following NNRTI-associated substitutions; K103N, Y181C, and G190A under clinically relevant concentrations.
A panel of 96 diverse clinical isolates containing NNRTI-associated mutations was evaluated for susceptibility to doravirine in the presence of 10 % foetal bovine serum. Clinical isolates containing the Y188L substitution or V106 substitutions in combination with A98G, H221Y, P225H, F227C or Y318F showed a greater than 100-fold reduced susceptibility to doravirine. Other substitutions yielded a fold change of 5-10 (G190S (5.7); K103N/P225H (7.9), V108I/Y181C (6.9), Y181V (5.1)). The clinical relevance of a 5-10 fold change reduction in susceptibility is unknown.
Treatment emergent doravirine resistance associated substitutions may confer cross-resistance to efavirenz, rilpivirine, nevirapine, and etravirine. Of the 8 subjects who developed high level doravirine resistance in the pivotal studies, 6 had phenotypic resistance to EFV and nevirapine, 3 to rilpivirine, and 3 had partial resistance to etravirine based on the Monogram Phenosense assay.
Lamivudine: Cross-resistance has been observed among NRTIs. The M184I/V lamivudine resistance substitution confers resistance to emtricitabine. Lamivudine-resistant HIV-1 mutants were also cross resistant to didanosine (ddI). In some subjects treated with zidovudine plus didanosine, isolates resistant to multiple RT inhibitors, including lamivudine, have emerged.
Tenofovir disoproxil: Cross-resistance has been observed among NRTIs. The K65R substitution in HIV-1 RT selected by tenofovir is also selected in some HIV-1 infected patients treated with abacavir or didanosine. HIV-1 isolates with the K65R substitution also showed reduced susceptibility to emtricitabine and lamivudine. Therefore, cross-resistance among these NRTIs may occur in patients whose virus harbours the K65R substitution. The K70E substitution selected clinically by tenofovir disoproxil results in reduced susceptibility to abacavir, didanosine, emtricitabine, lamivudine, and tenofovir. HIV-1 isolates from patients (n = 20) whose HIV-1 expressed a mean of 3 zidovudine associated RT amino acid substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) showed a 3.1-fold decrease in the susceptibility to tenofovir. Subjects whose virus expressed an L74V RT substitution without zidovudine resistance-associated substitutions (n = 8) had reduced response to tenofovir disoproxil. Limited data are available for patients whose virus expressed a Y115F substitution (n = 3), Q151M substitution (n = 2), or T69 insertion (n = 4) in HIV-1 RT, all of whom had a reduced response in clinical trials.
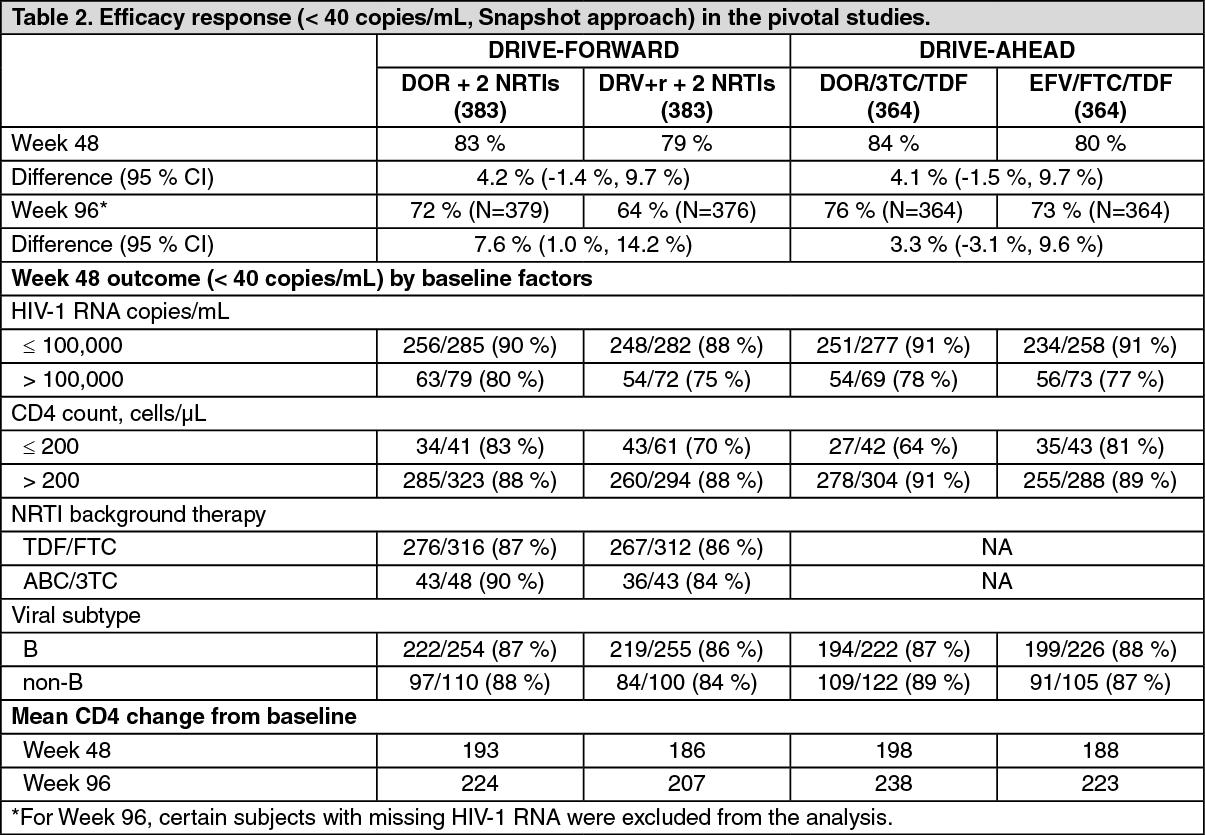
Clinical experience: Treatment-naïve adult subjects: The efficacy of doravirine is based on the analyses of 96-week data from two randomised, multicentre, double-blind, active controlled Phase 3 trials, (DRIVE-FORWARD and DRIVE-AHEAD) in antiretroviral treatment-naïve, HIV-1 infected subjects (n = 1494). Refer to Resistance as previously mentioned for NNRTI substitutions that were part of exclusion criteria.
In DRIVE-FORWARD, 766 subjects were randomised and received at least 1 dose of either doravirine 100 mg or darunavir + ritonavir 800+100 mg once daily, each in combination with emtricitabine/tenofovir disoproxil (FTC/TDF) or abacavir/lamivudine (ABC/3TC) selected by the investigator. At baseline, the median age of subjects was 33 years (range 18 to 69 years), 86 % had CD4+ T cell count greater than 200 cells per mm3, 84 % were male, 27 % were non-white, 4 % had hepatitis B and/or C virus co-infection, 10 % had a history of AIDS, 20 % had HIV-1 RNA greater than 100,000 copies per mL, 13 % received ABC/3TC and 87 % received FTC/TDF; these characteristics were similar between treatment groups.
In DRIVE-AHEAD, 728 subjects were randomised and received at least 1 dose of either doravirine/lamivudine/tenofovir disoproxil fumarate 100/300/300 mg (DOR/3TC/TDF) or efavirenz/emtricitabine/tenofovir disoproxil fumarate (EFV/FTC/TDF) once daily. At baseline, the median age of subjects was 31 years (range 18-70 years), 85 % were male, 52 % were non-white, 3 % had hepatitis B or C co-infection, 14 % had a history of AIDS, 21 % had HIV-1 RNA > 100,000 copies per mL, and 12 % had CD4+ T cell count < 200 cells per mm3; these characteristics were similar between treatment groups.
Week 48 and 96 outcomes for DRIVE-FORWARD and DRIVE-AHEAD are provided in Table 2. The doravirine-based regimens demonstrated consistent efficacy across demographic and baseline prognostic factors. (See Table 2.)
 Click on icon to see table/diagram/image
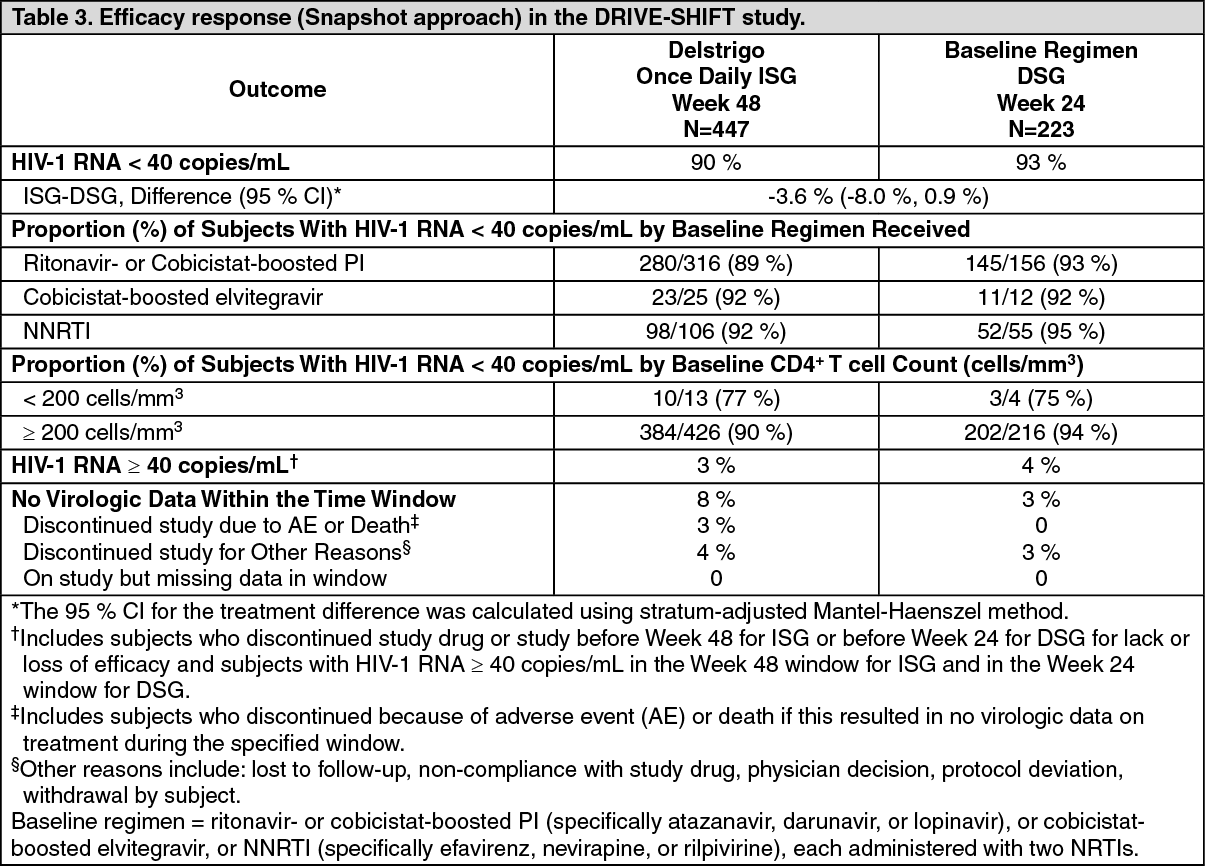
Click on icon to see table/diagram/imageVirologically suppressed adult subjects: The efficacy of switching from a baseline regimen consisting of two nucleoside reverse transcriptase inhibitors in combination with a ritonavir- or cobicistat-boosted PI, or cobicistat-boosted elvitegravir, or an NNRTI to Delstrigo was evaluated in a randomised, open-label trial (DRIVE-SHIFT), in virologically suppressed HIV-1 infected adults. Subjects must have been virologically suppressed (HIV-1 RNA < 40 copies/mL) on their baseline regimen for at least 6 months prior to trial entry, with no history of virologic failure, and a documented absence of RT substitutions conferring resistance to doravirine, lamivudine and tenofovir (see Resistance as previously mentioned). Subjects were randomised to either switch to Delstrigo at baseline [N = 447, Immediate Switch Group (ISG)], or stay on their baseline regimen until Week 24, at which point they switched to Delstrigo [N = 223, Delayed Switch Group (DSG)]. At baseline, the median age of subjects was 43 years, 16 % were female, and 24 % were non-white.
In the DRIVE-SHIFT trial, an immediate switch to Delstrigo was demonstrated to be non-inferior at Week 48 compared to continuation of the baseline regimen at Week 24 as assessed by the proportion of subjects with HIV-1 RNA < 40 copies/mL. Treatment results are shown in Table 3. Consistent results were seen for the comparison at study Week 24 in each treatment group. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDiscontinuation due to adverse events: In DRIVE-AHEAD, a lower proportion of subjects who discontinued due to an adverse event by Week 48 was seen for the Delstrigo group (3.0 %) compared with the EFV/FTC/TDF group (6.6 %).
Paediatric population: The efficacy of DOR/3TC/TDF was evaluated in an open-label, single-arm trial in HIV-1 infected paediatric patients 12 to less than 18 years of age (IMPAACT 2014 (Protocol 027)).
At baseline, the median age of subjects was 15 years (range: 12 to 17), 58 % were female, 78 % were Asian and 22 % were Black, and the median CD4+ T-cell count was 713 cells per mm3 (range: 84 to 1,397). After switching to DOR/3TC/TDF, 95 % (41/43) of virologically-suppressed subjects remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 24 and 93 % (40/43) remained suppressed (HIV-1 RNA < 50 copies/mL) at week 48.
The European Medicines Agency has deferred the obligation to submit the results of studies with Delstrigo in one or more subsets of the paediatric population in treatment of human immunodeficiency virus-1 (HIV-1) infection, as per Paediatric Investigation Plans (PIP) decision in the granted indication. See Dosage & Administration for information on paediatric use.
Pharmacokinetics: Single-dose administration of one doravirine/lamivudine/tenofovir disoproxil tablet to healthy subjects (N = 24) under fasted conditions provided comparable exposures of doravirine, lamivudine, and tenofovir to administration of doravirine tablets (100 mg) plus lamivudine tablets (300 mg) plus tenofovir disoproxil fumarate tablets (300 mg). The administration of a single Delstrigo tablet with a high-fat meal to healthy subjects resulted in a 26 % increase in doravirine C24, while AUC and Cmax were not significantly affected. Lamivudine Cmax decreased by 19 % with a high fat meal, while AUC was not significantly affected. Tenofovir Cmax decreased by 12 % and AUC increased by 27 % with a high fat meal. These differences in pharmacokinetics are not clinically relevant.
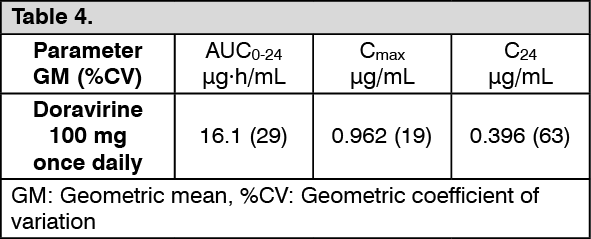
Doravirine: The pharmacokinetics of doravirine were studied in healthy subjects and HIV-1-infected subjects. Doravirine pharmacokinetics are similar in healthy subjects and HIV-1-infected subjects. Steady state was generally achieved by Day 2 of once daily dosing, with accumulation ratios of 1.2 to 1.4 for AUC0-24, Cmax, and C24. Doravirine steady state pharmacokinetics following administration of 100 mg once daily to HIV-1 infected subjects, based on a population pharmacokinetics analysis are provided as follows. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAbsorption: Following oral dosing, peak plasma concentrations are achieved 2 hours after dosing. Doravirine has an estimated absolute bioavailability of approximately 64 % for the 100 mg tablet.
Distribution: Based on administration of an IV microdose, the volume of distribution of doravirine is 60.5 L. Doravirine is approximately 76 % bound to plasma proteins.
Biotransformation: Based on in vitro data, doravirine is primarily metabolised by CYP3A.
Elimination: Doravirine: Doravirine has a terminal half-life (t1/2) of approximately 15 hours. Doravirine is primarily eliminated via oxidative metabolism mediated by CYP3A4. Biliary excretion of unchanged medicinal product may contribute to the elimination of doravirine, but this elimination route is not expected to be significant. Excretion of unchanged medicinal product via urinary excretion is minor.
Lamivudine: Following oral administration, lamivudine is rapidly absorbed and extensively distributed. After multiple-dose oral administration of lamivudine 300 mg once daily for 7 days to 60 healthy subjects, steady-state Cmax (Cmax,ss) was 2.04 ± 0.54 microgram per mL (mean ± SD) and the 24-hour steady-state AUC (AUC24,ss) was 8.87 ± 1.83 mcg·hour per mL. Binding to plasma protein is low. Approximately 71 % of an intravenous dose of lamivudine is recovered as unchanged medicinal product in the urine. Metabolism of lamivudine is a minor route of elimination. In humans, the only known metabolite is the trans-sulphoxide metabolite (approximately 5 % of an oral dose after 12 hours). In most single-dose trials in HIV-1 infected subjects, or healthy subjects with serum sampling for 24 hours after dosing, the observed mean elimination half-life (t½) ranged from 5 to 7 hours. In HIV-1-infected subjects, total clearance was 398.5 ± 69.1 mL/min (mean ± SD).
Tenofovir disoproxil fumarate: Following oral administration of a single 300 mg dose of tenofovir disoproxil fumarate to HIV-1-infected subjects in the fasted state, Cmax was achieved in one hour. Cmax and AUC values were 0.30 ± 0.09 micrograms per mL and 2.29 ± 0.69 μg·hr per mL, respectively. The oral bioavailability of tenofovir from tenofovir disoproxil in fasted subjects is approximately 25 %. Less than 0.7 % of tenofovir binds to human plasma proteins in vitro over the range of 0.01 to 25 micrograms per mL. Approximately 70-80 % of the intravenous dose of tenofovir is recovered as unchanged medicinal product in the urine within 72 hours of dosing. Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion with a renal clearance in adults with CrCl greater than 80 mL per minute of 243.5 ± 33.3 mL per minute (mean ± SD). Following oral administration, the terminal half-life of tenofovir is approximately 12 to 18 hours. In vitro studies have determined that neither tenofovir disoproxil nor tenofovir are substrates for the CYP450 enzymes.
Renal impairment: Doravirine: Renal excretion of doravirine is minor. In a study comparing 8 subjects with severe renal impairment to 8 subjects without renal impairment, the single dose exposure of doravirine was 31 % higher in subjects with severe renal impairment. In a population pharmacokinetic analysis, which included subjects with CrCl between 17 and 317 mL/min, renal function did not have a clinically relevant effect on doravirine pharmacokinetics. No dose adjustment is required in patients with mild, moderate or severe renal impairment. Doravirine has not been studied in patients with end-stage renal disease or in patients undergoing dialysis (see Dosage & Administration).
Lamivudine: Studies with lamivudine show that plasma concentrations (AUC) are increased in patients with renal dysfunction due to decreased clearance. Based on the lamivudine data, Delstrigo is not recommended for patients with CrCl of < 50 mL/min.
Tenofovir disoproxil fumarate: Pharmacokinetic parameters of tenofovir were determined following administration of a single dose of tenofovir disoproxil fumarate 300 mg to 40 non-HIV infected adult subjects with varying degrees of renal impairment defined according to baseline CrCl (normal renal function when CrCl > 80 mL/min; mild with CrCl = 50-79 mL/min; moderate with CrCl = 30-49 mL/min and severe with CrCl = 10-29 mL/min). Compared with subjects with normal renal function, the mean (% CV) tenofovir exposure increased from 2,185 (12 %) ng·h/mL in subjects with CrCl > 80 mL/min to respectively 3,064 (30 %) ng·h/mL, 6,009 (42 %) ng·h/mL and 15,985 (45 %) ng·h/mL in subjects with mild, moderate, and severe renal impairment.
The pharmacokinetics of tenofovir in non-haemodialysis adult subjects with CrCl < 10 mL/min and in subjects with end-stage renal disease managed by peritoneal or other forms of dialysis have not been studied.
Hepatic impairment: Doravirine: Doravirine is primarily metabolised and eliminated by the liver. There was no clinically relevant difference in the pharmacokinetics of doravirine in a study comparing 8 subjects with moderate hepatic impairment (classified as Child-Pugh score B primarily due to increased encephalopathy and ascites scores) to 8 subjects without hepatic impairment. No dose adjustment is required in patients with mild or moderate hepatic impairment. Doravirine has not been studied in subjects with severe hepatic impairment (Child-Pugh score C) (see Dosage & Administration).
Lamivudine: The pharmacokinetic properties of lamivudine have been determined in subjects with moderate to severe hepatic impairment. Pharmacokinetic parameters were not altered by diminishing hepatic function. Safety and efficacy of lamivudine have not been established in the presence of decompensated liver disease.
Tenofovir disoproxil fumarate: The pharmacokinetics of tenofovir following a 300 mg dose of tenofovir disoproxil fumarate have been studied in healthy subjects with moderate to severe hepatic impairment. No clinically relevant differences in tenofovir pharmacokinetics were observed between subjects with hepatic impairment and healthy subjects.
Paediatric population: Mean doravirine exposures were similar in 54 paediatric patients aged 12 to less than 18 years and weighing at least 35 kg who received doravirine or doravirine/lamivudine/tenofovir disoproxil IMPAACT 2014 (Protocol 027) relative to adults following administration of doravirine or doravirine/lamivudine/tenofovir disoproxil. Exposures of lamivudine and tenofovir in paediatric subjects following the administration of doravirine/lamivudine/tenofovir disoproxil were similar to those in adults following administration of lamivudine and tenofovir disoproxil (Table 5). (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageElderly: Although a limited number of subjects aged 65 years and over has been included (n = 36), no clinically relevant differences in the pharmacokinetics of doravirine have been identified in subjects at least 65 years of age compared to subjects less than 65 years of age in a Phase 1 trial or in a population pharmacokinetic analysis. The pharmacokinetics of lamivudine and tenofovir have not been studied in subjects older than 65 years. No dose adjustment is required.
Gender: No clinically relevant pharmacokinetic differences have been identified between men and women for doravirine, lamivudine, and tenofovir.
Race: Doravirine: No clinically relevant racial differences in the pharmacokinetics of doravirine have been identified based on a population pharmacokinetic analysis of doravirine in healthy and HIV-1-infected subjects.
Lamivudine: There are no significant or clinically relevant racial differences in pharmacokinetics of lamivudine.
Tenofovir disoproxil: There were insufficient numbers from racial and ethnic groups other than Caucasian to adequately determine potential pharmacokinetic differences among these populations following the administration of tenofovir disoproxil.
Toxicology: Preclinical safety data: Reproductive toxicity: Doravirine: Reproduction studies with orally administered doravirine have been performed in rats and rabbits at exposures approximately 9 times (rats) and 8 times (rabbits) the exposure in humans at the recommended human dose (RHD) with no effects on embryo-foetal (rats and rabbits) or pre/postnatal (rats) development. Studies in pregnant rats and rabbits showed that doravirine is transferred to the foetus through the placenta, with foetal plasma concentrations of up to 40 % (rabbits) and 52 % (rats) that of maternal concentrations observed on gestation Day 20.
Doravirine was excreted into the milk of lactating rats following oral administration, with milk concentrations approximately 1.5 times that of maternal plasma concentrations.
Lamivudine: Lamivudine was not teratogenic in animal studies but there were indications of an increase in early embryonic deaths in rabbits at relatively low systemic exposures, comparable to those achieved in humans. A similar effect was not seen in rats even at very high systemic exposure.
Tenofovir disoproxil: Reproductive toxicity studies in rats and rabbits showed no effects on mating, fertility, pregnancy or foetal parameters. However, tenofovir disoproxil reduced the viability index and weight of pups in a peri-postnatal toxicity study at maternally toxic doses.
Carcinogenesis: Doravirine: Long-term oral carcinogenicity studies of doravirine in mice and rats showed no evidence of carcinogenic potential at estimated exposures up to 6 times (mice) and 7 times (rats) the human exposures at the RHD.
Lamivudine: Long-term carcinogenicity studies with lamivudine in mice and rats showed no evidence of carcinogenic potential at exposures up to 12 times (mice) and 57 times (rats) the human exposures at the RHD.
Tenofovir disoproxil: Oral carcinogenicity studies in rats and mice only revealed a low incidence of duodenal tumours at an extremely high-dose in mice. These tumours are unlikely to be of relevance to humans.
Mutagenesis: Doravirine: Doravirine was not genotoxic in a battery of in vitro or in vivo assays.
Lamivudine: Lamivudine was mutagenic in an L5178Y mouse lymphoma assay and clastogenic in a cytogenetic assay using cultured human lymphocytes. Lamivudine was not mutagenic in a microbial mutagenicity assay, in an in vitro cell transformation assay, in a rat micronucleus test, in a rat bone marrow cytogenetic assay, and in an assay for unscheduled DNA synthesis in rat liver.
Tenofovir disoproxil: Tenofovir disoproxil was mutagenic in the in vitro mouse lymphoma assay and negative in an in vitro bacterial mutagenicity test (Ames test). In an in vivo mouse micronucleus assay, tenofovir disoproxil was negative when administered to male mice.
Impairment of fertility: Doravirine: There were no effects on fertility, mating performance or early embryonic development when doravirine was administered to rats at up to 7 times the exposure in humans at the RHD.
Lamivudine: Lamivudine did not affect male or female fertility in rats.
Tenofovir disoproxil: Reproductive toxicity studies in rats and rabbits showed no effects on mating, fertility, pregnancy or foetal parameters.
Repeat dose toxicity: Doravirine: Administration of doravirine in animal toxicity studies was not associated with toxicity.
Lamivudine: Administration of lamivudine in animal toxicity studies at high doses was not associated with any major organ toxicity. At the highest dosage levels, minor effects on indicators of liver and kidney function were seen together with occasional reductions in liver weight. The clinically relevant effects noted were a reduction in red blood cell count and neutropenia.
Tenofovir disoproxil: Findings in repeat-dose toxicity studies in rats, dogs and monkeys at exposure levels greater than or equal to clinical exposure levels and with possible relevance to clinical use included kidney and bone changes and a decrease in serum phosphate concentration. Bone toxicity was diagnosed as osteomalacia (monkeys) and reduced bone mineral density (BMD) (rats and dogs). The bone toxicity in young adult rats and dogs occurred at exposures ≥ 5-fold the exposure in paediatric or adult patients; bone toxicity occurred in juvenile infected monkeys at very high exposures following subcutaneous dosing (≥ 40-fold the exposure in patients). Findings in the rat and monkey studies indicated that there was a substance related decrease in intestinal absorption of phosphate with potential secondary reduction in BMD.