


 Sign Out
Sign Out




Bempedoic acid: Bempedoic acid is an adenosine triphosphate-citrate lyase (ACL) inhibitor that lowers low-density lipoprotein cholesterol (LDL-C) by inhibition of cholesterol synthesis in the liver. ACL is an enzyme upstream of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase in the cholesterol biosynthesis pathway. Bempedoic acid and its active metabolite, ESP15228, require coenzyme A (CoA) activation by very long-chain acyl-CoA synthetase 1 (ACSVL1) to ETC-1002-CoA and ESP15228-CoA, respectively. ACSVL1 is expressed primarily in the liver. Inhibition of ACL by ETC-1002-CoA results in decreased cholesterol synthesis in the liver and lowers LDL-C in blood via upregulation of low-density lipoprotein receptors.
Ezetimibe: Ezetimibe reduces blood cholesterol by inhibiting the absorption of cholesterol by the small intestine. The molecular target of ezetimibe has been shown to be the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is involved in the intestinal uptake of cholesterol and phytosterols. Ezetimibe localizes at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver. This causes a reduction of hepatic cholesterol stores and an increase in LDL receptors, resulting in clearance of cholesterol from the blood.
Pharmacodynamics: Administration of bempedoic acid and ezetimibe in combination with maximally tolerated statins, with or without other lipid-modifying agents, decreases LDL-C, non-high-density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (apo B), and total cholesterol (TC) in patients with hyperlipidemia.
Cardiac Electrophysiology: A QT trial has been conducted for bempedoic acid. At a dose of 240 mg (1.3 times the approved recommended dose), bempedoic acid does not prolong the QT interval to any clinically relevant extent.
The effect of ezetimibe or Nustendi on QT interval has not been evaluated.
Clinical Studies: The efficacy of Nustendi was investigated in a single, multi-center, randomized, double-blind, placebo-controlled, parallel group trial that enrolled 301 patients with heterozygous familial hypercholesterolemia, established atherosclerotic cardiovascular disease, or multiple risk factors for cardiovascular disease on maximally tolerated statin therapy. The efficacy of Nustendi in patients with multiple risk factors for cardiovascular disease has not been established.
Study 1 (NCT03337308) was a 4-arm, 12-week trial that assessed the efficacy of Nustendi in 301 patients randomized 2:2:2:1 to receive either Nustendi (180 mg of bempedoic acid and 10 mg of ezetimibe) (n = 86), bempedoic acid 180 mg (n = 88), ezetimibe 10 mg (n = 86), or placebo (n = 41) once daily as add-on to maximally tolerated statin therapy. Patients were stratified by cardiovascular risk and baseline statin intensity. Patients on simvastatin 40 mg per day or higher and patients taking non-statin lipid-lowering therapy (including fibrates, niacin, bile acid sequestrants, ezetimibe, and PCSK9 inhibitors) were excluded from the trial.
Overall, the mean age at baseline was 64 years (range: 30 to 87 years), 50% were ≥65 years old, 50% were women, 12% Hispanic, 81% White, 17% Black, and 1% Asian. Sixty-two percent (62%) of patients had clinical atherosclerotic cardiovascular disease (ASCVD) and/or a diagnosis of heterozygous familial hypercholesterolemia (HeFH). The mean baseline LDL-C was 149.7 mg/dL. At the time of randomization, 65% of patients were receiving statin therapy and 35% were receiving high-intensity statin therapy.
The primary efficacy outcome measure of the study was the percent change from baseline to Week 12 in LDL-C. The difference between Nustendi and placebo in mean percent change in LDL-C from baseline to Week 12 was -38% (95% CI: -47%, -30%; p < 0.001). High-density lipoprotein (HDL) and triglycerides (TG) were examined as exploratory endpoints and were not included in the statistical hierarchy. The difference between Nustendi and placebo in mean percent change from baseline to Week 12 was -5% for HDL and median percent change from baseline to Week 12 was -11% for TG. The maximum LDL-C lowering effect was observed at Week 4. For additional results, see Table 1.
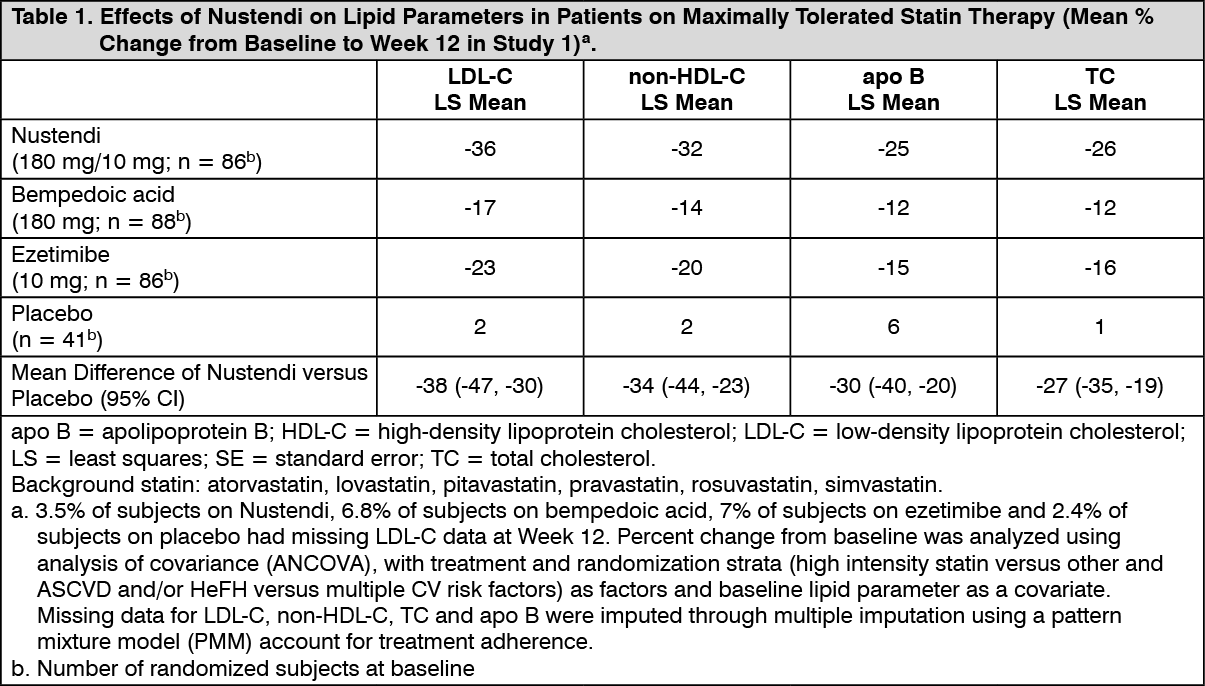 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageExamination of age, gender, and race subgroups did not identify differences in response to Nustendi among these subgroups in any of the trials.
Bempedoic Acid: In two 52-week trials that included 3009 adult patients with heterozygous familial hypercholesterolemia or established atherosclerotic cardiovascular disease on maximally tolerated statin therapy, the difference between bempedoic acid and placebo in mean percent change in LDL-C from baseline to Week 12 was -17% to -18%. Bempedoic acid also significantly lowered non-HDL-C (-13%), apo B (-12% to -13%), and TC (-11%) compared with placebo.
Ezetimibe: Ezetimibe Added to On-going Statin Therapy: In a multicenter, double-blind, placebo-controlled, 8-week study, 769 patients with primary hyperlipidemia, known coronary heart disease or multiple cardiovascular risk factors who were already receiving statin monotherapy, but who had not met their NCEP ATP II target LDL-C goal, were randomized to receive either ezetimibe or placebo in addition to their on-going statin therapy.
Ezetimibe, added to on-going statin therapy, significantly lowered TC (-17%), LDL-C (-25%), apo B (-19%), non-HDL-C (-23%), and TG (-14%), and increased HDL-C (+3%) relative to baseline and compared with a statin administered alone. LDL-C reductions induced by ezetimibe were generally consistent across all statins.
Ezetimibe Initiated Concurrently with a Statin: In four, multicenter, double-blind, placebo-controlled, 12-week trials, in 2382 hyperlipidemic patients, ezetimibe or placebo was administered alone or with various doses of atorvastatin, simvastatin, pravastatin, or lovastatin. When all patients receiving ezetimibe with a statin were compared to all those receiving the corresponding statin alone, ezetimibe significantly lowered LDL-C (ezetimibe + all atorvastatin doses [-56%] versus all atorvastatin doses alone [-44%]; ezetimibe + all simvastatin doses [-51%] versus all simvastatin doses alone [-36%]; ezetimibe + all pravastatin doses [-39%] versus all pravastatin doses alone [-25%]; ezetimibe + all lovastatin doses [-40%] versus all lovastatin doses alone [-25%]). LDL-C reductions induced by ezetimibe were generally consistent across all statins.
Pharmacokinetics: Absorption: Nustendi: The bioavailability of Nustendi tablets was similar relative to that from the individual tablets, co-administered. Maximum plasma concentration (Cmax) values for bempedoic acid and its active metabolite (ESP15228) were similar between formulations, but ezetimibe-glucuronide and ezetimibe Cmax values were approximately 22% and 13% lower, respectively, for Nustendi relative to the individual tablets, co-administered. Given a similar overall extent of ezetimibe-glucuronide and ezetimibe exposure (as measured by AUC), a 22% lower Cmax is unlikely to be clinically significant.
Bempedoic acid: Following single oral administration of Nustendi (180 mg of bempedoic acid and 10 mg of ezetimibe), mean (± SD) Cmax and AUC of bempedoic acid were 12.6 (± 2.80) μg/mL and 202 (± 43.4) μg·hr/mL, respectively; the median time to maximum concentration (Tmax) was 3.0 hours. Following multiple-dose administration of bempedoic acid monotherapy, the steady-state maximum plasma concentration (Cmax) and AUC at 180 mg/day were 20.6 ± 6.1 μg/mL and 289.0 ± 96.4 μg·h/mL, respectively. Bempedoic acid steady-state pharmacokinetics were generally linear over a range of >60 mg to 220 mg (approximately 33% to 122% of the recommended dosage of 180 mg daily). There were no time-dependent changes in bempedoic acid pharmacokinetics following repeat administration at the recommended dosage, and bempedoic acid steady-state was achieved after 7 days. The mean accumulation ratio was approximately 2.3-fold.
The steady-state Cmax and AUC of the active metabolite (ESP15228) of bempedoic acid were 2.8 ± 0.9 μg/mL and 51.2 ± 17.2 μg·h/mL, respectively. ESP15228 likely made a minor contribution to the overall clinical activity of bempedoic acid based on systemic exposure, relative potency, and pharmacokinetic properties.
Ezetimibe: After a single dose of Nustendi to fasted adults, mean ± SD ezetimibe Cmax of 3.56 ± 1.90 ng/mL were attained with a median Tmax of 5 hr. Ezetimibe-glucuronide mean Cmax values of 107 ± 46 ng/mL were achieved with a median Tmax of 1 hr. For ezetimibe monotherapy, there was no substantial deviation from dose proportionality between 5 mg and 20 mg (0.5- to 2-fold the recommended dosage). The absolute bioavailability of ezetimibe cannot be determined, as the compound is virtually insoluble in aqueous media suitable for injection.
Effect of Food: Nustendi: After the administration of Nustendi with a high-fat, high-calorie breakfast in healthy subjects, the AUC for bempedoic acid and ezetimibe were comparable to the fasted state. Compared to the fasted state, the fed state resulted in 30% and 12% reductions in Cmax and 2-hour and 2.5-hour delays in median time to attain maximum concentration (Tmax) of bempedoic acid and ezetimibe, respectively. For ezetimibe-glucuronide, a 12% and 42% decrease in AUC and Cmax, respectively, were observed under fed relative to fasted conditions.
This effect of food is not considered to be clinically meaningful.
Distribution: Bempedoic acid: The bempedoic acid apparent volume of distribution (V/F) was 18 L. Plasma protein binding of bempedoic acid, its glucuronide and its active metabolite, ESP15228, were 99.3%, 98.8% and 99.2%, respectively. Bempedoic acid does not partition into blood cells.
Ezetimibe: Ezetimibe and ezetimibe-glucuronide are highly bound (>90%) to human plasma proteins.
Elimination: Bempedoic acid: The steady-state clearance (CL/F) of bempedoic acid was 11.2 mL/min after once-daily dosing; renal clearance of unchanged bempedoic acid represented less than 2% of total clearance. The mean ± SD half-life for bempedoic acid in humans was 21 ± 11 hours at steady-state.
Ezetimibe: Both ezetimibe and ezetimibe-glucuronide are eliminated from plasma with a half-life of approximately 22 hours for both.
Metabolism: Bempedoic acid: The primary route of elimination for bempedoic acid is through metabolism to the acyl glucuronide. Bempedoic acid is also reversibly converted to an active metabolite (ESP15228) based on aldo-keto reductase activity observed in vitro from human liver. Mean plasma AUC metabolite/parent drug ratio for ESP15228 following repeat-dose administration was 18% and remained constant over time. Both bempedoic acid and ESP15228 are converted to inactive glucuronide conjugates in vitro by UGT2B7. Bempedoic acid, ESP15228 and their respective conjugated forms were detected in plasma with bempedoic acid accounting for the majority (46%) of the AUC0-48h and its glucuronide being the next most prevalent (30%). ESP15228 and its glucuronide represented 10% and 11% of the plasma AUC0-48h, respectively.
Ezetimibe: Ezetimibe is primarily metabolized in the small intestine and liver via glucuronide conjugation with subsequent biliary and renal excretion. Minimal oxidative metabolism has been observed in all species evaluated.
In humans, ezetimibe is rapidly metabolized to ezetimibe-glucuronide. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10% to 20% and 80% to 90% of the total drug in plasma, respectively. Plasma concentration-time profiles exhibit multiple peaks, suggesting enterohepatic recycling.
Excretion: Bempedoic acid: Following single oral administration of 240 mg of bempedoic acid (1.3 times the approved recommended dose), approximately 70% of the total dose (bempedoic acid and its metabolites) was recovered in urine, primarily as the acyl glucuronide conjugate of bempedoic acid, and approximately 30% was recovered in feces. Less than 5% of the administered dose was excreted as unchanged bempedoic acid in feces and urine combined.
Ezetimibe: Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe (ezetimibe + ezetimibe-glucuronide) accounted for approximately 93% of the total radioactivity in plasma. Approximately 78% and 11% of the administered radioactivity were recovered in the feces and urine, respectively, over a 10-day collection period. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Ezetimibe was the major component in feces and accounted for 69% of the administered dose, while ezetimibe-glucuronide was the major component in urine and accounted for 9% of the administered dose.
Specific Populations: Patients with Renal Impairment: Bempedoic acid: Pharmacokinetics of bempedoic acid was evaluated in a single-dose pharmacokinetic study in subjects with varying degrees of renal function. The mean bempedoic acid AUC in subjects with mild renal impairment (n = 8) were 1.5-fold higher compared to those with normal renal function (n = 6). Relative to those with normal renal function, mean bempedoic acid AUCs were higher in patients with moderate (n = 5) or severe (n = 5) renal impairment by 2.3-fold and 2.4-fold, respectively.
A population pharmacokinetic analysis was performed on pooled data from all clinical trials (n = 2261) to further evaluate the effects of renal function on the steady-state AUC of bempedoic acid. Compared to patients with normal renal function, the mean bempedoic acid exposures were higher in patients with mild or moderate renal impairment by 1.4-fold (90% CI: 1.3, 1.4) and 1.9-fold (90% CI: 1.7, 2.0), respectively. These differences were not clinically significant. Clinical studies of bempedoic acid did not include patients with severe renal impairment (eGFR <30 mL/min/1.73 m2) or patients with ESRD on dialysis [see Renal Impairment under Precautions].
Ezetimibe: After a single 10 mg dose of ezetimibe in patients with severe renal disease (n = 8; mean CrCl ≤30 mL/min/1.73 m2), the mean AUC for total ezetimibe, ezetimibe-glucuronide, and ezetimibe increased approximately 1.5-fold, compared to healthy subjects (n = 9). No dosage adjustment is necessary for the ezetimibe component. However, there is limited experience with bempedoic acid in patients with severe renal impairment [see Renal Impairment under Precautions].
Patients with Hepatic Impairment: Nustendi is not recommended in patients with moderate or severe hepatic impairment due to the unknown effects of the increased exposure to ezetimibe [see Hepatic Impairment under Precautions].
Bempedoic acid: The pharmacokinetics of bempedoic acid and its metabolite (ESP15228) was studied in patients with normal hepatic function or mild or moderate hepatic impairment (Child-Pugh A or B) following a single dose (n = 8/group). Compared to patients with normal hepatic function, the bempedoic acid mean Cmax and AUC were decreased by 11% and 22%, respectively, in patients with mild hepatic impairment and by 14% and 16%, respectively, in patients with moderate hepatic impairment. Compared to patients with normal hepatic function, the ESP15228 mean Cmax and AUC were decreased by 13% and 23%, respectively, in patients with mild hepatic impairment and by 24% and 36%, respectively, in patients with moderate hepatic impairment. This is not expected to result in lower efficacy.
Bempedoic acid was not studied in patients with severe hepatic impairment (Child-Pugh C).
Ezetimibe: After a single 10 mg dose of ezetimibe, the mean AUC for total ezetimibe increased approximately 1.7-fold in patients with mild hepatic impairment (Child-Pugh A), compared to healthy subjects. The mean AUC values for total ezetimibe and ezetimibe increased approximately 3- to 4-fold and 5- to 6-fold, respectively, in patients with moderate (Child-Pugh B) or severe hepatic impairment (Child-Pugh C). In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic impairment, the mean AUC for total ezetimibe and ezetimibe increased approximately 4-fold on Day 1 and Day 14 compared to healthy subjects.
Other Specific Populations: Bempedoic acid: The pharmacokinetics of bempedoic acid were not affected by age, gender, race, or weight.
Ezetimibe: Geriatrics: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were about 2-fold higher in older (≥65 years) healthy subjects compared to younger subjects [see Use in the Elderly under Precautions].
Gender: In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were slightly higher (<20%) in women than in men.
Race: The pharmacokinetics of ezetimibe is not affected by race.
Drug Interaction Studies: Bempedoic acid: Cytochrome P450 Substrates: In vitro metabolic interaction studies suggest that bempedoic acid, as well as its active metabolite and glucuronide forms, are not metabolized by and do not interact with cytochrome P450 enzymes.
Transporter-mediated Drug Interactions: In vitro drug interaction studies suggest bempedoic acid, as well as its active metabolite and glucuronide form, are not substrates of commonly characterized drug transporters with the exception of bempedoic acid glucuronide, which is an OAT3 substrate. Bempedoic acid weakly inhibits OAT3 at high multiples of clinically relevant concentrations, and bempedoic acid and its glucuronide weakly inhibit OATP1B1, and OATP1B3 at clinically relevant concentrations. Bempedoic acid weakly inhibits OAT2 in vitro, which is likely the mechanism responsible for minor elevations in serum creatinine and uric acid [see Clinical Trials Experience under Adverse Reactions]. Inhibition of OAT2 by bempedoic acid may also potentially increase plasma concentrations of medicinal products that are substrates of OAT2.
Probenecid: Administration of bempedoic acid 180 mg with steady-state probenecid resulted in a 1.7- and a 1.2-fold increase in bempedoic acid AUC and Cmax, respectively. AUC and Cmax for bempedoic acid active metabolite (ESP15228) were increased 1.9- and 1.5-fold, respectively. These elevations are not clinically meaningful and do not impact dosing recommendations.
Statins: The pharmacokinetic interactions between bempedoic acid (at systemic exposure relevant to the indicated ASCVD population) and simvastatin 20 mg, atorvastatin 10 mg, pravastatin 40 mg, and rosuvastatin 10 mg were evaluated in clinical trials.
Simvastatin: Administration of simvastatin 20 mg with 240 mg of bempedoic acid or 40 mg with 180 mg of bempedoic acid in healthy subjects at steady-state resulted in approximately 2-fold (91% for 20 mg and 96% for 40 mg) and 1.5-fold (54% for 20 mg and 52% for 40 mg) increases in simvastatin acid AUC and Cmax, respectively [see Interactions].
Pravastatin: Administration of pravastatin 40 mg with steady-state bempedoic acid 240 mg in healthy subjects resulted in 99% (2-fold) and 104% (2-fold) increases in pravastatin acid AUC and Cmax, respectively [see Interactions].
Atorvastatin and Rosuvastatin: Elevations of 1.7-fold in AUC of atorvastatin and rosuvastatin and/or their major metabolites were observed, suggesting a weak interaction. These elevations were generally within the individual statin exposures and do not impact dosing recommendations.
Warfarin: In vitro studies indicate that bempedoic acid is not an inhibitor or inducer of CYP2C9. Because warfarin is primarily eliminated through CYP2C9, its pharmacokinetics is not expected to be altered by bempedoic acid.
Other: Bempedoic acid had no effect on the pharmacokinetics of metformin or the oral contraceptive Ortho-Novum 1/35.
Ezetimibe: Ezetimibe had no significant effect on a series of probe drugs (caffeine, dextromethorphan, tolbutamide, and IV midazolam) known to be metabolized by cytochrome P450 (1A2, 2D6, 2C8/9 and 3A4) in a "cocktail" study of twelve healthy adult males. This indicates that ezetimibe is neither an inhibitor nor an inducer of these cytochrome P450 isozymes, and it is unlikely that ezetimibe will affect the metabolism of drugs that are metabolized by these enzymes.
Cyclosporine: Administration of ezetimibe with cyclosporine (75-150 mg BID) resulted in a 2.4- and a 2.9-fold increase in total ezetimibe AUC and Cmax, respectively [see Interactions].
Fibrates: Administration of ezetimibe with fenofibrate (200 mg QD for 14 days) resulted in a 1.48- and a 1.64-fold increase in total ezetimibe AUC and Cmax, respectively. Administration with gemfibrozil (600 mg BID for 7 days) resulted in a 1.64- and 1.91-fold increase in total ezetimibe AUC and Cmax, respectively [see Interactions].
Cholestyramine: Administration of ezetimibe with cholestyramine (4 g BID for 14 days) resulted in a 55% and a 4% decrease in total ezetimibe AUC and Cmax, respectively [see Interactions].
No clinically meaningful pharmacokinetic interaction was observed following co-administration of ezetimibe with aluminum & magnesium hydroxide combination antacid, cimetidine, glipizide, lovastatin, pravastatin, atorvastatin, rosuvastatin, fluvastatin, simvastatin, digoxin, ethyl estradiol/levonorgestrel, and warfarin.
Toxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Bempedoic acid: Bempedoic acid was negative for mutagenicity in an in vitro Ames assay and negative for clastogenicity in the in vitro human lymphocyte chromosome aberration assay. Bempedoic acid was negative in both in vivo mouse micronucleus and in vivo rat bone marrow micronucleus/liver comet assay. In a 2-year rat carcinogenicity study, Wistar rats were given oral doses of bempedoic acid at 3, 10 and 30 mg/kg/day. An increased incidence of liver hepatocellular adenomas and hepatocellular adenomas combined with carcinomas, thyroid gland follicular cell adenoma and follicular cell adenomas combined with carcinomas, and pancreatic islet cell adenomas combined with carcinomas were observed in male rats at the dose of 30 mg/kg/day (exposure equivalent to the maximum recommended human dose (MRHD), based on AUC). In a 2-year mice carcinogenicity study, CD-1 mice were given oral doses of bempedoic acid at 25, 75 and 150 mg/kg/day. Bempedoic acid-related increases in the incidence of liver hepatocellular adenomas, hepatocellular carcinomas and hepatocellular adenomas combined with carcinomas in male mice were observed at 75 and 150 mg/kg/day (exposures equivalent to the MRHD). Observations of liver and thyroid tumors are consistent with PPAR alpha agonism in rodents. The human relevance of pancreatic islet cell tumor findings is unknown.
In fertility and early embryofetal development study in rats, bempedoic acid was given orally to male and female rats at 10, 30 and 60 mg/kg/day. Males were dosed for 28 days prior to mating and females were dosed 14 days prior to mating through gestation day 7. No adverse effects on fertility were observed in females in the absence of maternal toxicity. No effects were observed on male fertility outcomes, but decreases in sperm counts were observed at 60 mg/kg/day (9 times the MRHD).
Ezetimibe: A 104-week dietary carcinogenicity study with ezetimibe was conducted in rats at doses up to 1500 mg/kg/day (males) and 500 mg/kg/day (females) (approximately 20 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). A 104-week dietary carcinogenicity study with ezetimibe was also conducted in mice at doses up to 500 mg/kg/day (>150 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). There were no statistically significant increases in tumor incidences in drug-treated rats or mice.
No evidence of mutagenicity was observed in vitro in a microbial mutagenicity (Ames) test with Salmonella typhimurium and Escherichia coli with or without metabolic activation. No evidence of clastogenicity was observed in vitro in a chromosomal aberration assay in human peripheral blood lymphocytes with or without metabolic activation. In addition, there was no evidence of genotoxicity in the in vivo mouse micronucleus test.
In oral (gavage) fertility studies of ezetimibe conducted in rats, there was no evidence of reproductive toxicity at doses up to 1000 mg/kg/day in male or female rats (approximately 7 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe).